
Research Areas
Extending Oxidative Chemistry beyond the Constraints of the Protein Environment
On this page:
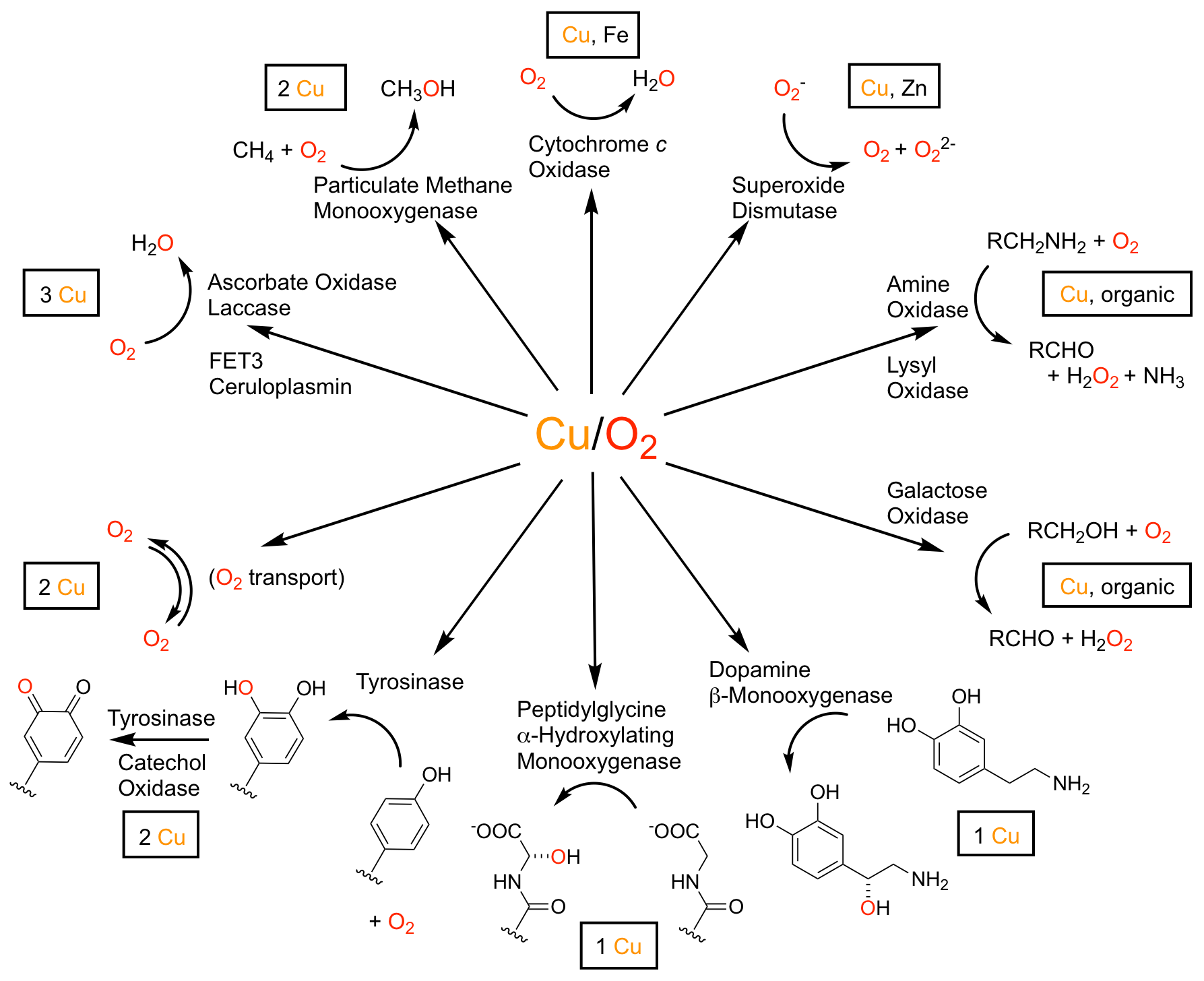
Copper-Dioxygen Chemistry
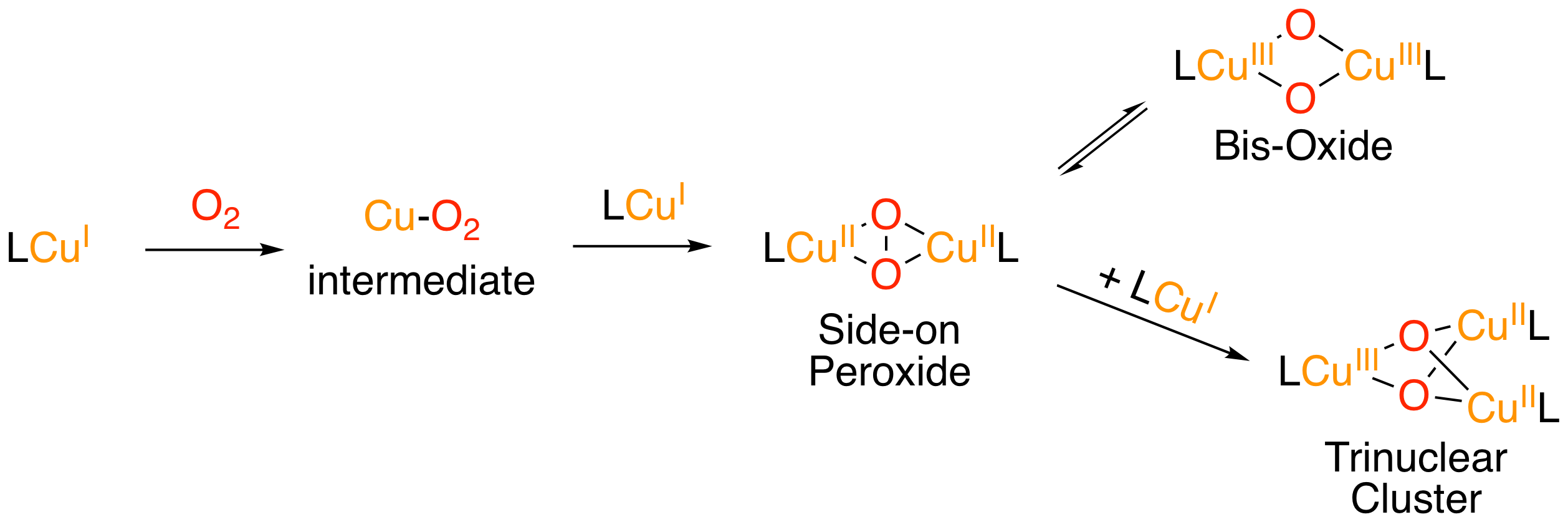
The ability of copper-containing enzymes to activate molecular dioxygen in energy metabolism and in the selective oxidation of biological substrates far surpasses the control and efficiency achieved by experimental chemists. The binuclear copper enzyme tyrosinase activates molecular dioxygen to a μ-η2:η2-peroxide dicopper(II) species in the catalytic, regioselective hydroxylation of tyrosine to an o-catechol - the first biosynthetic step invoked in pigment formation and oxidative fruit browning. The intimate mechanistic details of this remarkable transformation have eluded biochemical investigation, largely due to the transient nature of intermediates formed in the reaction in aqueous conditions. Another pressing academic challenge and economic opportunity is the high-efficiency reduction of dioxygen to water. The trinuclear copper enzyme laccase performs this transformation with a lower overpotential (ca. 100 mV) than any other known catalyst. As in the binuclear, spectroscopic data diverge on the assignment of the intermediates in this mechanism. The use of small-molecule active-site models in the bioinorganic chemistry of copper has yielded a wealth of information, previously unobtainable through enzymatic studies, concerning structure-reactivity relationships and the potential role of different oxidation states of copper in biosynthetic pathways.
Surface Immobilized Oxidation Catalysts
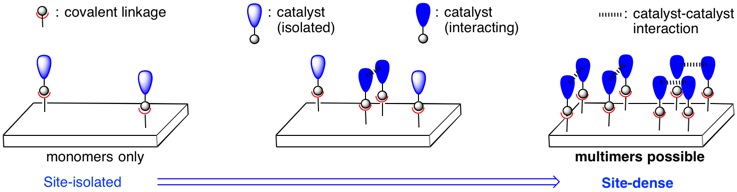
A comprehensive picture of homogeneous transition-metal catalyzed reactions is often difficult to achieve due to an incomplete understanding of the speciation of the active catalyst and intermediates in the reaction process. In the solution phase, unrestricted interactions of catalyst molecules can lead to complexes of unknown composition through multimerization or transformation of the ligand scaffold. These processes obscure insight into the mechanism of catalysis and may also contribute to deactivation pathways.
Covalent immobilization of discrete molecular complexes to a bulk material, such as mesoporous silica (SBA-15), enables investigation of outstanding questions in homogeneous catalysis, most notably catalyst speciation and the effects of catalyst-catalyst interactions. Tuning the distribution of tethered catalyst molecules allows exploration of molecular species under the limiting cases of "site-isolated" and "site-dense", as well as intermediate cases. In the site-isolated case, catalyst molecules are spatially far apart; catalyst-catalyst interactions are disabled, and speciation is straightforward under such conditions. Conversely, in the site-dense case, catalyst molecules are spatially close, and catalyst-catalyst interactions can occur readily. Comparisons of reactivity under these limiting heterogenized conditions and homogeneous conditions can clarify salient mechanistic details and illuminate possible strategies for improvements to catalysis.